
FDA자문위원회는 가속승인상태의 알츠하이머치료제 '레켐비'(레카네맙)의 정식승인전환을 만장일치로 권고했다.
자문위는 9일 에자이와 바이오젠의 알츠하이머 치료제 '레켐비'의 확증 3상 Clarity AD(NCT03887455)이 임상적 이점에 대한 증거를 제공하는지 여부에 대한 질의에 6:0 만장일치의 찬반 투표를 통해 승인을 권고하는 의견을 냈다.
FDA는 자문위의 의견을 기반으로 오는 7월 6일 정식승인 전환여부를 결정할 예정으로 1월 6일 가속승인 6개월만에 정식승인까지 확보할 가능성이 높아졌다.

에자이가 제시한 임상결과에 따르면 투약 18개월차 기준 알츠하이머 질환의 진행을 27%(치매임상평가척도/CDR-SB 차이 0.48) 늦췄으며 강력한 아밀로이트 플라크 제거 효과 등을 제시했다.
아밀로이드 관련 영상이상(ARIA) 부작용 관련 3건의 사망사건 이슈와 관련해 약물과의 연관성이 유력한 2건을 식별하고 투약관련 관리의 강화 필요성이 제기됐다.
이외 자문위를 통해 심각한 부작용 사례가 집중된 하위그룹군 아포지단백E(ApoE) ε4/4 동형접합군의 경우의 유일하게 위약군 대비 1차 평가변수 기준 레켐비가 선호되지 않는 환자군임이 확인됐다.
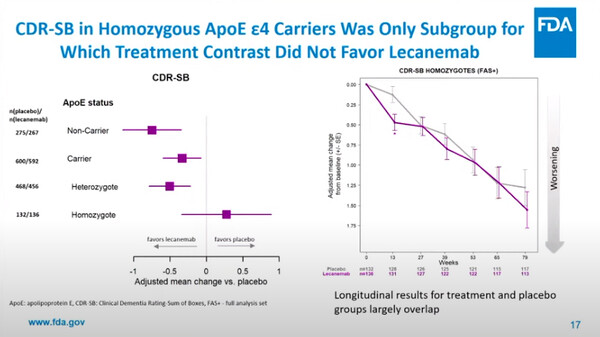
다만 CDR-SB의 이점은 없었으나 2차 평가변수인 AD평가척도(ADAS-cog14)등에서 다른 하위군에 비해 낮지만 일부 치료효과를 제시하고 있다.
임상 투약군의 22%에 해당하는 해당 군에서 심각한 부작용사례가 집중되고 실제 투약효과가 크지 않은 만큼 투약 대상에서 유지/제외할지 또는 해당환자군에 대해서만 별도의 경고가 추가되지 여부는 정해지지 않았다. 내달 FDA 승인결정시 최종 확정될 전망이다.
