한미약품이 개발하고 스펙트럼사가 FDA 가속승인을 추진중인 항암제 포지오티닙에 상업화 일정에 먹구름이 드리웠다.
FDA는 오는 22일(현지시간) 개최예정인 자문위원회을 앞두고 공개한 브리핑 문서를 통해 승인 신청 기반이되는 ZENITH20 임상결과가 승인을 뒷받침하기에는 충분하지 않다는 의견을 피력했다.
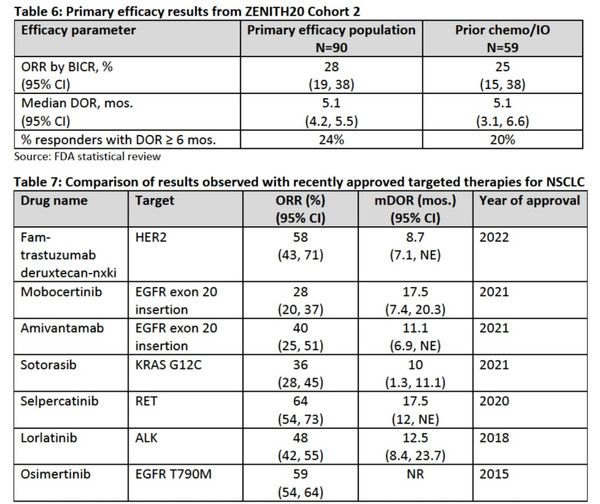
포지오티닙은 이전 국소치료를 받은 HER2 엑손 20입 삽입 변이 비소세포폐암을 적응증으로 지난해 11월 1일 FDA 가속승인 신청서를 제출했으며 승인목표예정일은 11월 24일로 잡혀있다.
환자군에서 보여준 객관적반응율은 28%(95% CI: 19, 38), DOR 중앙값 5.1개월(95% CI: 4.2, 5.5)였다.
FDA는 가속승인 용량으로 제안된 1일 1회 16mg 투약군에서 용량감소를 경험한 경험한 환자의 비율이 57%이며 3,4등급의 이상반응을 경험한 환자군이 85%에 달했다고 설명했다. 이상반응의 경우 용량을 줄임으로 그 비율이 낮아질 수 있다고 덧붙였다.
이어 이같이 용량감소를 경험한 환자가 많아 기존 치료법 대비 더 많은 이점을 제공한다고 예측 또는 평가하기에는 어려움이 있으며 이상반응을 고려할 경우 위험대비 이점을 갖고 있다고 평가를 내리기는 시기상조라고 진단했다.
또한 가속승인 후 확증임상은 1일 2회 8mg투약 방식의 다른 용량을 제안하고 있으며 2026년 완료돼, 임상효능의 확인이 지연될 수 있다는 점도 우려사항으로 꼽았다. 즉 가속승인과 확증임상 용량의 불일치 문제를 지적했다.
일단 FDA는 포지오티닙에 대해 부정적인 입장을 제시함에 따라 22일 개최되는 자문위에서 승인 권고 의견을 확보할 가능성이 낮아졌다.